
La epilepsia es un problema de salud pública a nivel
mundial. Se considera que afecta a 50 millones de personas en todo el
mundo. En Estados Unidos se presenta en 5-10 personas de cada mil. En el
grupo pediátrico tiene una incidencia de 72-86 casos por cada cien mil
niños menores de nueve años de edad. Únicamente en 30% de los casos se
logrará identificar una etiología específica, correspondiendo en los
menores de 15 años a trastornos asociados a lesiones cerebrales (trauma,
encefalopatía hipóxico-isquémica, anomalías cerebrales y/o infecciones
congénitas).1,2,3,4,5,6.
El término Convulsión se refiere a la alteración de la conducta
resultado de la actividad anormal y excesiva de un grupo de neuronas
cerebrales.1,3.
Epilepsia se define como la presencia de dos o más convulsiones no
provocadas, esto es, sin que se haya logrado identificar algún factor
que se asocie a ellas (fiebre, infección del sistema nervioso central,
trauma de cráneo, etc.). Debe considerarse que la epilepsia es una
condición clínica caracterizada por fenómenos clínicos recurrentes, en
la mayoría de las veces autolimitados. Muchos trastornos episódicos
caracterizados por movimientos involuntarios y/o alteración o pérdida de
la conciencia no están asociados a cambios electroencefalográficos
paroxísticos.1,3,7.
Clasificación
Una de las prioridades iniciales al evaluar a un niño con
convulsiones es determinar el tipo de crisis y si es posible catalogarlo
dentro de algún síndrome epiléptico.1,2,3.
Las convulsiones se dividen en las que afectan sólo a una región
cerebral (parciales), y aquellas que involucran a ambos hemisferios
cerebrales desde su inicio (generalizadas). Las parciales se subdividen
en las que no logran comprometer el estado de conciencia (simples) y en
aquellas en las que si hay compromiso de ella (complejas); en algunos
casos, estos tipos de crisis pueden desembocar en crisis tónico-clónicas
secundariamente generalizadas.1,2,3,8.
Las crisis que afectan la corteza motora consistirán de actividad
clónica de la cara, brazo o pierna. Las crisis parciales simples
usualmente duran menos de un minuto y no se asocian a sintomatología
post-ictal; sin embargo, cuando las crisis son prolongadas y comprometen
el área motora se desarrollará cierta debilidad transitoria en los
grupos musculares afectados (Parálisis de Todd). Los síntomas psíquicos
usualmente ocurren como componentes de una crisis parcial
compleja.1,2,3,8.
 |
Síndromes epilépticos pediátricos
Síndrome de West: Una de las encefalopatías epilépticas
edad-dependientes. Se presenta en hasta 25 de cada 100,000 nacidos vivos
en Estados Unidos y en Europa. Inicia entre los 4 y 7 meses de edad.
Únicamente en 75% de los casos se logrará identificar una causa; dentro
de los trastornos más frecuentemente asociados están las malformaciones
congénitas, asfixia perinatal y la esclerosis tuberosa. Las crisis
clásicas corresponden a los llamados espasmos infantiles (flexores,
extensores y/o mixtos), de duración muy breve, y que pueden asociarse en
salvas (grupos). El trazo electroencefalográfico característico es la
hipsarritmia (punta-onda lenta de muy alto voltaje, prácticamente
durante todo el registro, con frecuentes períodos decrementales de
voltaje). Dentro de las modalidades terapéuticas utilizadas, la
vigabatrina es de las que se asocia a mejores resultados, sobre todo en
los casos secundarios a esclerosis tuberosa. En nuestro medio aún sigue
siendo válido el uso de ácido valproico como fármaco de primera línea,
pudiendo llegar a alcanzarse en algunos casos dosis entre 100-300 mg/Kg.
El pronóstico es reservado, sobre todo en aquellos niños en los que se
logra identificar una causa.1,2,9,10.
Síndrome de Lennox-Gastaut: Inicia entre los 3-5 años de edad. Hasta
en 60% de los casos se logra identificar la etiología. En 20% de los
casos se documenta el antecedente de haber cursado con Síndrome de West.
La tríada clásica conocida comprende convulsiones (tónicas, atónicas,
mioclónicas y ausencias atípicas), complejos punta-onda lenta (1.5-2.5
Hz) en el electroencefalograma y retraso mental (más del 90% a la edad
de cinco años). Las convulsiones son refractarias al uso de medicamentos
(ácido valproico, clonacepam, lamotrigina, topiramato, etc.); siendo
posible en la actualidad recurrir al uso de modalidades terapéuticas no
convencionales en buen número de casos (dieta cetogénica, estimulación
vagal, cirugía).1,2,11.
Síndrome de Janz: Es un trastorno que se hereda como rasgo autosómico
dominante (6p21.3). Su inicio se ubica entre los 7-13 años de edad.
Afecta a ambos géneros por igual. Se asocia a crisis tónico-clónicas, de
ausencia y/o mioclónicas, de predominio al despertar; las cuales
frecuentemente son desencadenadas por la deprivación de sueño y la
ingestión de alcohol. Electroencefalográficamente muestran paroxismos de
punta y polipunta-onda generalizadas, simétricas, de 3.5-6 Hz, de mayor
voltaje en regiones frontocentrales, altamente sensibles al
fotoestímulo. El ácido valproico es el tratamiento de primera línea,
logrando control absoluto en hasta el 75% de los casos.1,2,3.
Crisis de Ausencia: Inicia entre los 4-8 años de edad. Se hereda como
rasgo autosómico dominante. Los niños afectados son completamente
normales desde el punto de vista neurológico. Las crisis duran de 5-30
segundos, pudiendo llegar a ser hasta 100-150 por día. Clásicamente se
presentan como fijación de la mirada y parpadeo, pero pueden asociarse a
componentes atónico, mioclónico y/o automatismos motores. No se asocian
a fenómeno post-ictal. El trazo electroencefalográfico se considera
patognómonico (complejos punta-onda simétricos, sincrónicos, de 3-4 Hz),
muy sensible a la hiperventilación. No se requiere el realizar estudios
de neuroimagen. El tratamiento de elección, a pesar del paso del
tiempo, continua siendo etosuximida y/o ácido valproico, logrando el
control absoluto de las crisis hasta en 80% de los casos; pudiendo ser
corroborado a través de estudios electroencefalográficos. La
carbamacepina no está indicada, ya que exacerba las crisis, en algunas
ocasiones desencadenando estado de ausencia. Los pacientes que llegan a
presentar crisis tónico-clónicas generalizadas y/o mioclónicas durante
su evolución serán de difícil control o progresarán a epilepsia
mioclónica juvenil.1,2,3.
Síndrome de Rasmussen: Inicia entre el primero y los 14 años de edad.
Se caracteriza por crisis parciales simples de tipo clónico, continuas,
que persisten por semanas o meses (epilepsia parcial continua),
seguidas de hemiparesia y hemiatrofia cerebral. En aproximadamente la
mitad de los casos es precedida por un evento infeccioso o inflamatorio.
Se han documentado infiltrados linfocíticos perivasculares, sobre todo a
nivel cortical. Los eventos convulsivos son refractarios al empleo de
antiepilépticos. El uso de inmunoglobulinas se ha reportado como eficaz.
El tratamiento definitivo consiste en la realización de hemisferectomía
anatómica.1,3.
 |
Diagnóstico
El obtener una historia clínica cuidadosa y detallada
permanece como la base para poder llegar a un diagnóstico
exacto.1,3,12,13.
El estudio electroencefalográfico es paroxístico en un tercio de los
pacientes con epilepsia; su positividad puede incrementarse al 90%
empleando procedimientos de activación (hiperventilación,
fotoestimulación, deprivación de sueño). El uso de electrodos no
convencionales está justificado, sobre todo, en crisis parciales
complejas (esfenoidales, nasoetmoidales, faringeos).1,3,8,12.
La presencia de descargas electroencefalográficas interictales no
establece el diagnóstico de epilepsia; pudiendo ser encontradas como
hallazgo incidental en hasta 2.4% de los niños en edad
escolar.1,3,8,12,13.
Las crisis parciales simples del lóbulo límbico frecuentemente no tienen correlato electroencefalográfico.1,3,12.
 |
Diagnóstico diferencial
Hasta un tercio de la población referida a las clínicas
altamente especializadas en el manejo de epilepsia, en realidad
corresponden a una entidad diferente a ésta, siendo en buena parte de
las veces condiciones de naturaleza benigna.1,3,7.
Hasta un tercio de los síncopes no llegan a ser reconocidos o son erróneamente diagnosticados como convulsiones.1,3,7.
 |
Tratamiento
No debe llegar a medicarse a los niños que únicamente
presenten actividad epiléptica subclínica en el
electroencefalograma.1,3,8,12,13,14.
Antes de iniciar el uso de medicamentos debe procurarse el lograr un
adecuado entendimiento de la naturaleza de la enfermedad y de los
objetivos del tratamiento, así como el obtener el mayor grado de
cooperación por parte de la familia y del paciente.1,3,12,13,14,15,16.
Debe procurarse lograr el control clínico y electroencefalográfico de
las crisis sin llegar a comprometer el bienestar físico e intelectual
del paciente, así como intentar preservarle una adecuada vida familiar y
social.1,3,16.
Todos los anticonvulsivantes disponibles hasta la fecha no están exentos de presentar efectos adversos.1,3,14.
La oportunidad de lograr el éxito en el manejo de la epilepsia es
mayor cuando éste se inicia en los primeros dos años de la
enfermedad.1,6,14.
Aproximadamente 70-80% de los niños con epilepsia mostrarán una
respuesta satisfactoria al uso de anticonvulsivantes después de un
período de tiempo relativamente corto. Cuando nos enfrentamos con un
paciente que no responde en forma adecuada habrá que cuestionarnos
acerca de la certeza del diagnóstico, de si la condición del paciente
obedece a una entidad de carácter progresivo, de si el medicamento
empleado es el adecuado para un tipo de crisis en particular, de si la
dosis prescrita es la indicada y tomar en cuenta las posibles
interacciones medicamentosas que nos puedan entorpecer el mismo.1,6,14.
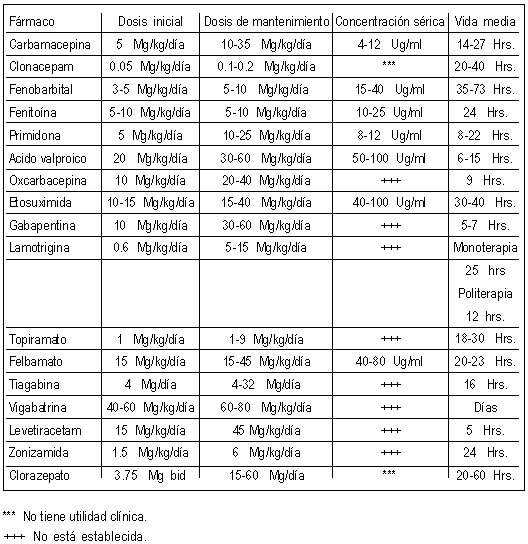
Las siguientes son indicaciones para obtener niveles séricos del o los anticonvulsivantes utilizados:
Verificar el apego del paciente
Persistencia de las crisis a pesar de utilizar un esquema terapéutico prescrito adecuadamente
Presencia o sospecha de efectos adversos
Presencia de descontrol de crisis en pacientes previamente bien controlados
Utilización de varios medicamentos que presenten interferencia, sean o no anticonvulsivantes
Empleo de medicamentos con un estrecho rango terapéutico cuando la edad
del paciente o la presencia de discapacidad física o mental es tal, que
las manifestaciones clínicas debidas a toxicidad sean difíciles o
imposibles de evaluar.
Pacientes muy pequeños, recién nacidos, debido a las frecuentes
oscilaciones en la concentración sérica que pueden condicionar
ineficacia o toxicidad antes de modificar el esquema utilizado,
especialmente cuando el medicamento empleado ha resultado ineficaz.1.
En la actualidad se encuentran disponibles importantes abordajes
terapéuticos no farmacológicos o no convencionales, como el uso de
vitaminas (piridoxina, biotina); el tratamiento dietético (dieta
cetogénica); el uso de inmunoglobulinas, esteroides y/o hormona
adrenocorticotrófica; y la estimulación vagal.1,6,12,14,17,18,19.
 |
Manejo quirúrgico
Lo más importante es identificar los casos que son
refractarios al manejo médico. En Estados Unidos se ha llegado a
considerar que aparecen 5,000 nuevos candidatos cada año, siendo en su
gran mayoría adultos.1,3,6,12,13,15,16.
 |
La cirugía puede mejorar el control de las crisis,
limitar la intensidad de las manifestaciones clínicas, disminuir los
efectos adversos de los antiepilépticos utilizados sobre la conducta y/o
la función cognoscitiva, limitar la propagación de áreas
epileptogénicas, evitar riesgos innecesarios a los pacientes (laborales,
etc.).1,8,11,12,15,16.
Pronóstico
El pronóstico de la epilepsia, en niños, va directamente
relacionado con el tipo de síndrome epiléptico identificado. La tasa de
recaída para aquellos que han alcanzado la remisión es del 20-63%,
siendo más frecuentes durante la fase de supresión de los
anticonvulsivantes o en el primero o segundo años después de haberlos
retirado. La epilepsia que inicia después del período de lactante y
antes de la adolescencia presenta mayores probabilidades de
remisión.1,3,6,12,20.
La historia de crisis neonatales incrementa el riesgo de recurrencia y la posibilidad de desarrollar crisis parciales.1,12,13.
Los siguientes son factores que nos permitirán predecir la adecuada remisión de las crisis:
Evaluación neurológica normal
Función intelectual normal
Ausencia de lesión cerebral
Presencia de un solo tipo de crisis
Descontrol de crisis relativamente breve, así como una rápida y buena respuesta al tratamiento antiepiléptico prescrito
Inicio tardío de las crisis (después de los 3 a 4 años)
Baja frecuencia de crisis
Ausencia de crisis tónicas y/o atónicas, estado epiléptico, crisis parciales complejas
Electroencefalograma normal al inicio del tratamiento o lograr su normalización en el transcurso del mismo
Adecuado apego al tratamiento.1,6,8,12,13.
El pronóstico será mejor en los casos de epilepsia criptogénica y en los
niños con un adecuado desarrollo neuropsicomotor antes del inicio de
las crisis o en el momento de realizar la evaluación inicial.1,6,8,12,.
Los niños epilépticos sin lesión orgánica y con desarrollo
neuropsicomotor adecuado tienen el 85% de probabilidades de llegar a
estar libre de crisis.1,6,8,12.
El deterioro cognoscitivo y/o conductual está asociado a algunos
síndromes epilépticos en especial (Síndrome de West, Síndrome de
Lennox-Gastaut, epilepsia mioclónica severa, Síndrome de
Landau-Kleffner, punta onda lenta continua durante el sueño
lento).1,6,8,9,12,19.
Muchos pacientes epilépticos desarrollarán problemas de aprendizaje durante el transcurso de su enfermedad.1,8,12,14.
La mortalidad de los niños con epilepsia durante los primeros diez
años es de 5.7%; siendo mayor en aquellos casos que inician antes del
primer año de edad, en los de epilepsia sintomática y en los que
presentan espasmos infantiles.1,6,8,12,14.
BIBLIOGRAFÍA1.- Aicardi, J. Classification of epileptic seizures and epilepsies. In Epilepsy in children. Raven Press 1994. 9-17.
2.- Commision on classification and terminology of the International
League against Epilepsy: proposal for revised classification of
epilepsies and epileptic syndromes. Epilepsia 1989; 30:389-99.
3.- Chang, B. S. and D. H. Lowenstein. Epilepsy. N Engl J Med. 2003 Sept 349(13):1275-66.
4.- Dale, C.H. et al. Risk of unprovoked seizure after acute
symptomatic seizure: effect of status epilepticus. Ann Neurol 1998 Dec
44(6):908-12.
5.- Hauser, W.A. et al. Risk of recurrent seizure after two unprovoked seizures. N Engl J Med 1998 Feb 338 (7):429-434.
6.- Sisodiya, S.M. Surgery for malformation of cortical development causing epilepsy. Brain 2000 Jun 123 (6):1075-91.
7.- Aicardi, J. La Epilepsia como un trastorno no paroxístico. Acta Neuropediátrica 1996 2(4):248-58.
8.- Devinsky, O. Patients with refractory seizures. N Engl J Med 1999 May 340 (20):1565-70.
9.- Watanabe, K. West syndrome: etiological and prognostic aspects. Brain Dev 1998; 20:1-8.
10.- Heiskala, H. et al. West síndrome: individualized ACTH therapy. Brain Dev 1996; 18:456-60.
11.- Motte, J., et al. Lamotrigine for generalized seizures
associated with the Lennox-Gastaut syndrome. N Engl J Med 1997 Dec
337:187-92.
12.- Kwan, Patrick. Early identification of refractory epilepsy. N Engl J Med 2000 Feb 342 (5):314-19.
13.- Arroyo, S. Diagnóstico y tratamiento del paciente con epilepsia farmacorresistente. Neurología 1996 Feb 11(2):56-65.
14.- Dieter, S. Rational Polytherapy. Bailliere’s Clinical Neurology. 1996 Dec 5(4):757-63.
15.- Kral, T. et al. Outcome of epilepsy surgery in focal cortical dysplasia. J Neurol Neurosurg Psychiatry 2003 74:183-8.
16.- Wiebe, Samuel et al. A randomized, controlled trial of surgery
for temporal-lobe epilepsy. N Engl J Med 2000 Aug 345 (5):311-18.
17.- Hassan, A. M. et al. Ketogenic diet in the treatment of
refractory epilpesy in childhood. Pediatr Neurol 1999 21 (2):548-52.
18.- Freeman, J. M. et al. The efficacy of the ketogenic diet-1998: A
prospective evaluation of intervention in 150 children. Pediatrics 1998
Dec 102(6):1358-63.
19.- Nordli, D. R., et al. Experience with the ketogenic diet in infants. Pediatrics 2001 July 108(1).
20.- Tennison, M., et al. Discontinuing antiepileptic drugs in
children with epilepsy: a comparison of a six week and a nine-month
taper period. N Engl J Med 1994 May 330(20):1407-10.