Equivalencia terapéutica, generalidades y conceptos
medidas de control y evaluación específicas que guíen el proceso
regulatorio de aprobación de medicamentos por genéricos/copias y así,
asegurar buenas prácticas de intercambiabilidad, especialmente cuando se
trata de drogas de dosis crítica.
Antecedentes
La trayectoria de un producto innovador, desde su concepción hasta
que llega al consumidor es ardua, ya que la investigación y desarrollo
de innovaciones, requiere de una contribución multidisciplinaria para
alcanzar los objetivos terapéuticos.
Cada país tiene su propia entidad regulatoria que es responsable de
desarrollar políticas y lineamientos para regular la dispensación de
genéricos/copias, sin embargo, en algunos casos la sustitución es
obligatoria y es manejada por asuntos económicos o por presión política,
y en otros casos los genéricos/copias no han sido sometidos
notablemente en el mercado y su uso es opcional. El empuje y lanzamiento
de los genéricos es puramente económico y se asume que su uso puede
reducir significativamente los costos en salud. Adicionalmente, las
drogas genéricas son usualmente más baratas que los innovadores, pero
las diferencias en precio no siempre son enormes y el costo curativo del
fallo terapéutico o efectos tóxicos, pueden rápidamente cancelar
cualquier costo ahorrado por la sustitución genérica.
Se debe aceptar una sustitución siempre y cuando la seguridad y eficacia no esté comprometida.
En 1977 la FDA introdujo la regla 75/75 (2 drogas eran consideradas
bioequivalentes sí los parámetros de biodisponibilidad eran 75% a 125%
de la referencia para 75% de la población test (o diferir de no más de
+/- 25%)). Para drogas que demostraron mayor inter e intra-variabilidad
en parámetros farmacocinéticos, la regla del 70/70 fue aplicada, la cual
permitió un +/- 30% de variabilidad. Estas reglas fueron muy criticadas
por su arbitrariedad y falta de validez estadística, por lo que ya no
son usadas por la FDA. Luego, los lineamientos de la FDA y la OMS
tuvieron aceptación estadística similares para bioequivalencia: La
aprobación de genéricos/copias requieren análisis estadísticos para cada
parámetro de biodisponibilidad evaluado, el cual debe mostrar un 90% de
certeza dentro de un rango de 80% a 125% del producto de referencia.
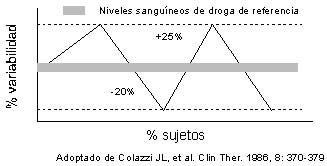
Las diferencias en ABC y Cmax entre el innovador y el genérico deber ser
considerablemente menor que –20% a +25%.
Sin embargo, bioequivalencia no necesariamente se traduce a
equivalencia terapéutica. Por ejemplo, un excipiente de una formulación
puede inhibir la unión de los receptores de la droga a la célula blanco,
o bien, tener efectos farmacológicos propios.
 |
Contenido:
La OMS ha recomendado a las Autoridades de Salud
requerir equivalencia terapéutica tanto para nuevos como para genéricos,
y para drogas críticas, sugiere que sean más estrictos y rigurosos en
los ensayos.
Debido a que no existen lineamientos solicitando los estándares de
aprobación de los genéricos, esto es considerado preferiblemente para
tener un acuerdo universal de criterios de aprobación y aceptación de
genéricos para simplificar y estandarizar los requisitos regulatorios en
todos los países especialmente en los de escasos recursos. Para este
fin la OMS ha sido consultada por la conferencia Internacional de
Autoridades Regulatorias para el desarrollo de lineamientos para
determinar la Equivalencia Terapéutica. Muchas entidades de Registro
fuera de USA son guiadas por lo requerido por la FDA. Las listas del
Libro Naranja (Orange Book de sus siglas en inglés) con drogas de varias
fuentes son listados con un sistema de códigos de dos letras:
A Terapéuticamente equivalente al innovador
B No Terapéuticamente equivalente al innovador
AB Indica que tiene problemas de bioequivalencia potenciales que tienen que ser resueltos
A+ Indica que ha sido una droga de referencia / A1, A2 Indica que hay dos o más referencias.
Hay muchas drogas listadas en el Libro Naranja para el cual una o
varias marcas tienen un rango de inequivalencia terapéutica, y hay aún
productos que han sido cambiados de A a B basados en la información
recibida después de la aprobación inicial, sugiriendo que los análisis
originales fueron insuficientemente strictos o fueron inaplicables a las
poblaciones de pacientes que recibieron la droga. En ciertas
instancias, la discrepancia terapéutica entre genéricos e innovadores ha
sido bastante seria para la FDA en revisar y demostrar nuevos
estándares, y luego retirar algunos genéricos/copias del mercado.
Para la mayoría de las drogas, los estándares regulatorios actuales
para demostrar bioequivalencia entre productos innovadores y
genérico/copias, son adecuados. Sin embargo, algunas categorías de
Productos de Dosis Crítica han sido identificados como difíciles para
establecer equivalencia terapéutica.
Para drogas de dosis críticas, aún los más pequeños cambios en
niveles de droga en plasma, pueden causar reacciones adversas en
eficacia clínica o toxicidad, y por lo tanto requieren intervención
médica, o bien se amenaza la vida.
Para la mayoría de drogas críticas, el problema no está en el uso de
genéricos o no, más bien, las dificultades tienden a aumentar cuando se
les cambia de la droga de la cual sí se han estabilizado.
El cambio indiscriminado por genéricos bajo los requisitos
regulatorios actuales de bioequivalencia puede dirigir a variaciones en
niveles de la droga a más de un 45%, lo cual es inaceptable en drogas de
dosis crítica. Además, el cambio requiere monitoreo sanguíneo, para
asegurar que los niveles terapéuticos de la droga son mantenidos. Los
médicos necesitarán tener un alto grado de sospecha o desconfianza en la
sustitución de genéricos, pues sino en vez de controlar la enfermedad
se atribuirá al cambio en el progreso de la enfermedad, interacciones de
la droga, no-cumplimiento por parte del paciente… y no, a la
sustitución del fármaco.
Por lo tanto, un fallo para detectar y compensar los niveles
plasmáticos inapropiados por ajuste de drogas puede comprometer el
beneficio clínico del tratamiento. Ejemplo de drogas críticas: litio,
ciclosporina, warfarina, fenitoína, levotiroxina, carbamazepina,
digoxina, quimodina, teofilina.
Tomaremos a la ciclosporina como un EJEMPLO: La ciclosporina es un
complejo lipofílico con absorción dependiente de la bilis, por lo que su
formulación es indispensable, además que cumplir con las demás
características. La inmunosupresión sub-óptima está asociada con el
incremento de incidencia de rechazo agudo y subsecuente, pérdida del
injerto, mientras que la ciclosporina en niveles arriba del rango
terapéutico aumenta el riesgo de fallo renal o nefrotoxicidad,
complicaciones infecciosas y linfomas, por lo que requiere
hospitalización e intervención médica costosa, ya que el paciente
regresa a diálisis. Por lo que ambos extremos son sumamente costosos y
son un riesgo para la vida del paciente. Claramente, con la
disponibilidad de órganos para trasplantes se han disminuido a nivel
mundial, un esfuerzo consciente debe ser hecho para evitar la necesidad
de un órgano adicional para retrasplantar debido a un fallo del producto
para proveer inmunosupresión dentro de la ventana terapéutica para
ciertos pacientes. Es muy importante que las Autoridades Regulatorias
evalúen la equivalencia terapéutica. Nuevas formulaciones deben ser
evaluadas clínicamente aún si la bioequivalencia con voluntarios sanos
es confirmada, pues aun en pacientes estables se pueden demostrar
diferentes resultados. Por lo tanto se debe tener mucho cuidado al
sustituir por genéricos/copias sobre todo si la bioequivalencia clínica
aún no ha sido adecuadamente demostrada.
Se podría argumentar que toda droga es de uso crítico, pero este
término lo que intenta es identificar drogas que están siendo usadas en
personas quienes están en alto riesgo (posiblemente trascendental) de
resultados negativos.
Ejemplo: Imipenem-cilastatina en pacientes con disfunción renal. Esta
droga se usa para tratar infecciones difíciles y tiene generalmente un
rango terapéutico amplio, sin embargo, es eliminado por vía renal. Y en
este paciente, la droga puede crear convulsiones (pues no hay
eliminación de la droga). Por lo que esta droga en este paciente es de
uso crítico, pues debe administrarse a una dosis correcta o espaciada
para lograr que el cuerpo tenga tiempo de eliminarlo y evitar toxicidad.
Ejemplo: La naturaleza no linear del metabolismo de la fenitoína,
grandes dosis de la droga puede resultar inicialmente un pequeño cambio
de concentración en el plasma, pero en cuanto se acerca a la curva de
dosis-concentración, un minúsculo aumento de dosis puede desproporcionar
el incremento de concentración en plasma, y generalmente provoca
toxicidad.
 |
Recomendaciones
El Centro de Salud: Debe educar al paciente sobre
fármacos genéricos/copias y debe incluir al paciente en las decisiones
de cambios de fármacos.
El farmacéutico: Debe informar al médico y al paciente cuando un
fármaco de dosis crítica sea cambiado. No se debería permitir la
sustitución sin la aprobación del médico y el paciente.
Médicos: Deben buscar información sobre la bioequivalencia de agentes
que recetan, cuando un médico informado se preocupa sobre el
mantenimiento de regímenes farmacológicos consistentes o sobre la
bioequivalencia de fármacos genéricos/copias, deberían ejercitar su
opción de solicitar que no se hagan sustituciones sin su consentimiento.
Los pacientes: Deben ser educados a identificar la forma del
medicamento recetado, y deben ser educados de que deben informar a su
médico si se les sustituyera un medicamento.
Autoridades Regulatorias: Debería requerir que la apariencia de todo
medicamento sea única y fácilmente identificable para permitir que los
pacientes distingan distintos fármacos.
Monitoreo: Debido a que las potenciales consecuencias que surgen a
partir de las diferencias en cuanto a biodisponibilidad o variabilidad
en cada sujeto con diferentes drogas críticas, los médicos deben
considerar el uso de monitoreo apropiado (niveles sanguíneos si fuera
necesario), siempre que un paciente sea cambiado de la droga utilizada.
Farmacovigilancia: El equipo de trabajadores de salud debe reportar
efectos adversos con medicamentos innovadores y genéricos/copias, a las
Autoridades de Salud, pacientes y, fabricantes con el fin de documentar
esta información. El médico debe considerar informar a otros miembros
del equipo.
Glosario:
· Producto Innovador: Una solicitud de registro de un
fármaco nuevo e innovador con toda la información relevante, que le
permita a la FDA evaluar y decidir si es aprobado o no el fármaco para
el mercado y uso humano.
· Producto Genérico/Copia: Es un fármaco que es comparable a un
innovador registrado (cuya patente ha expirado) o uno en una lista de
referencia en su aspectos de dosis, dosificación, forma, potencia, ruta
de administración, calidad, características de funcionamiento, y de su
proyectado uso.
· Equivalencia Farmacéutica: Se refiere a fármacos que contienen la
misma sustancia activa, la misma concentración, misma forma de
dosificación y se administra por la misma vía. Lleva a resultados
comparables in vitro.
· Biodisponibilidad: Expresa simultáneamente la velocidad e
intensidad de la disponibilidad (o absorción paso de sustancia activa a
través de una barrera fisiológica) del principio activo (llega a la
circulación general) en su sitio de acción, tal que puedan describirlo
los niveles plasmáticos.
· Bioequivalencia: Tienen una biodisponibilidad comparable cuando son administrados con la misma posología a los mismos sujetos.
· Equivalencia Terapéutica: Dos productos farmacéuticos son
equivalentes terapéuticamente si son farmacéuticamente equivalentes y
después de su administración en la misma dosis molar sus efectos, con
respecto a eficacia como a seguridad, serán esencialmente las mismas
como pueden derivarse de estudios apropiados.
· Intercambiabilidad: Es la habilidad de cambiar un paciente de una formulación del medicamento a otra.
· Prescripción/sustitución: La prescripción puede ser con nombre de
marca o como sustancia activa, si no quiere ser cambiado indicarlo en la
receta. El farmacéutico puede hacer la sustitución pero dependiendo del
tipo de droga debe informar tanto al médico como al paciente de dicha
sustitución.
· Drogas de Dosis Crítica: Son aquellas en las que un pequeño cambio
en dosis o concentración causa un cambio clínico significativo en
eficacia o toxicidad. Estas drogas tienen características en común:
- Margen terapéutico estrecho
- Amplia variabilidad intra e inter-pacientes
- Absorción errática o limitada
- Biodisponibilidad o disolución que depende de la formulación
- Consecuencias que ponen en riesgo la vida del paciente con
la administración de dosis sub-óptimas y supra-óptimas.
- Dosificación generalmente basada en peso (mg/kg.) o superficie
corporal (mg/m2)
- Monitoreo en sangre
Referencias:
1. http://www.fda.gov/cder/ob/default.htm
2. http://www.fda.gov/cder
3. http://www.fda.gov/medwatch/safety.htm
4.• http://www.fda.gov/cder/approval/index.htm
5. http://www.usp.org
6. http://www.who.int/medicines/library/qsm/manual-on-marketing/multisource-contents.html
7. Substitution of Critical-Dose Drugs: Issues, Analyses and Decision
Making. American Pharmaceutical Association, The National Professional
Society of Pharmacists. 2000:1-17.
8. Atholl Johnston Presentation. The Safe Use of Generic Formulations in
Transplantation. Reader in Clinical Pharmacology inSt Bartholomew’s and
the Royal London School of Medicine and Dentistry. London UK.
Publicaciones relacionadas
- El ultrasonido musculoesquelético y la reumatología
- Evaluación pronóstica del uso de octreótide 2 vrs. 5 días en sangrado gastrointestinal superior de origen variceal
- Ley de Murphy para Médicos
- Acerca de nosotros y nuestros servicios
- Acromegalia
- Adyuvancia extendida en Cáncer de mama
- Aprendiendo, Jorge Luis Borges
- Areas nasales por tomografia axial computarizada:
- Ascitis: Mecanismos de producción. Diagnóstico y Tratamiento Dr. J. Alberto Marin
- Asociación CRECE
- Aspectos filosóficos e históricos en la práctica de la medicina interna Casi todos los médicos tienen sus enfermedades favoritas Dr. Hugo Raúl Castro Salguero. Médico Internista, Oncólogo. HGE – IGSS. Instituto Nacional de Cancerología INCAN. Grupo Médico Ángeles
- Calidad de vida en pacientes con hepatitis crónica por virus c vírgenes a tratamiento
- Canal: grupo medico angeles en youtube
- Características de los pacientes diagnosticados con enfermedad de Hansen en los últimos diez años en el INDERMA, Dr. Peter Greenberg Cordero, Director Médico del INDERMA; Dra. Suzzette de León, Jefa de Unidad de Hansen del INDERMA; Dra. Helga María Sarti, Residente III del INDERMA
- Carcinoma Basocelular Superficial: reporte de un caso
- Carcinoma de células de Merkel
- Caso Interesante
- Casos de Autodiscusión
- Casos de autodiscusión 2
- Cinco casos de enfermos con fiebre de origen desconocido
- Colonoscopia virtual 3d tan sensible como la colonoscopia de fibra
- Consejos útiles para dejar de fumar
- Cuerpo editorial
- Curiosidades de la Ciencia y de la Vida
- Curiosidades de la Ciencia y de la Vida 2