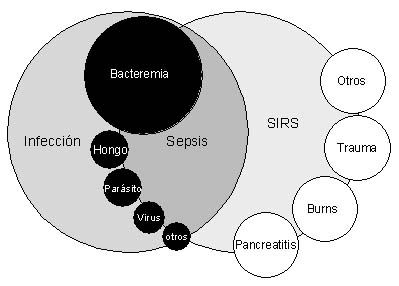
Patogenesis del Síndrome de Sepsis y Choque Séptico
La respuesta inflamatoria sistémica secundaria a
infección se denomina sepsis, es secundaria a una compleja respuesta del
huésped a la infección en la que interactúan el endotelio vascular, la
respuesta inflamatoria y la coagulación, que de no revertir evolucionan a
disfunción orgánica múltiple. A pesar de la identificación del síndrome
de respuesta anti-inflamatoria sistémica compensadora (SARC) que
incluye moléculas neutralizantes mediadoras, tales como receptores de
citocinas, y receptores antagonistas de citocinas. Estas moléculas
contrarrestan la excesiva liberación de las citocinas proinflamatorias,
uniéndose a las citocinas e interfiriendo con sus interacciones en
receptores celulares de membrana finales (1)
La liberación de endotoxinas o exotoxinas por las bacterias induce a
la activación de macrófagos los que sintetizan y liberan citocinas
proinflamatorias que inducen cambios a nivel endotelial, y modifican el
equilibrio procoagulante –anticoagulante, proceso que evoluciona a
obstrucción y mal funcionamiento de la microcirculación con disfunción
orgánica múltiple.
La respuesta inflamatoria es mediada por endotoxinas, activación del
sistema inmune, leucotrienos , componentes del complemento y citocinas,
las cuales son péptidos inmunorreguladores sintetizados y liberados por
los macrófagos y que interactúan sobre receptores localizados en
diferentes líneas celulares principalmente a nivel de linfocitos,
macrófagos, médula ósea y endotelio vascular. (2)
La respuesta inflamatoria guarda un estrecho equilibrio que es
mediado por citocinas proinflamatorias y antiinflamatorias; dentro de
las citocinas proinflamatorias podemos mencionar: el Factor de Necrosis
tumoral alfa (FNT alfa) Interleucina 1 (IL-1) Interleucina 2 (IL-2)
Interleucina 6 (IL-6) Interleucina 8 (IL-8) e Interferón gamma, que
tienen los siguientes efectos biológicos: Síntesis de óxido nítrico,
activación del factor nuclear Kappa Beta (FNKB), expresión del factor
tisular (FT), modulación del gen de expresión de trombomodulina,
activación de fibrinolisis, expresión de moléculas de adhesión
endotelial, activación de polimorfonucleares (PMN), fiebre, síntesis de
proteínas de fase aguda por el hígado, modificación del metabolismo
intermedio, así como maduración y diferenciación de células B y T y
megacariocitos. (3)
Este proceso condiciona el síndrome de respuesta inflamatoria
sistémica (SIRS) que de no controlarse evoluciona a falla orgánica
múltiple
La respuesta proinflamatoria es antagonizada por la liberación de
citocinas antiinflamatorias como son la Interleucina 4 (IL-4)
Interleucina 10 (IL-10) y antagonistas de receptores de citocinas, que
inhiben la expresión de moléculas de adhesión, del FT y los efectos
vasculares mediados por óxido nítrico, leucotrienos y radicales libres
de oxigeno, además de modular la función de linfocitos T, macrófagos y
la síntesis de inmunoglobulinas y citocinas, lo que constituye el
síndrome de respuesta antiinflamatoria compensadora (SRAC). (1)
El endotelio vascular conforma la interfase entre sangre y tejidos
conformada por células endoteliales que tiene funciones biológicas
fundamentales como:
Modulación de la coagulación, Regulación del flujo microvascular,
Expresión de moléculas de adhesión , Regulación de la migración de
células a los tejidos y Modulación del tono vascular, dentro de las
cuales la modulación de la coagulación es una función fundamental del
endotelio que presenta una marcada tendencia anticoagulante, la cual
tiene como finalidad el mantener el flujo microvascular por los
siguientes mecanismos:
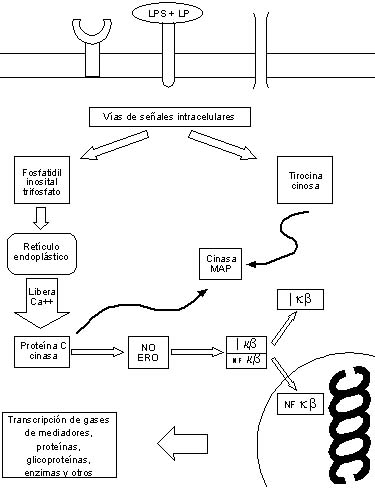 |
1. Expresión de trombomodulina, la cual tiene como
función la fijación de la trombina , así como el incremento de la
afinidad de ésta a la proteína C. Una vez activada la proteína C por
trombomodulina y unida a su cofactor (proteína S) inactiva
catalíticamente a los factores V y VII.
2. A través de proteinglicanos como el heparán sulfato que se
encuentra en la superficie endotelial, se potencia la acción de
inhibidores de coagulación como son la Antitrombina III (ATIII) y el
inhibidor del factor tisular.
3. Síntesis y liberación del activador del factor tisular del plasminógeno.
4. Inhibición de la agregación plaquetaria mediada por prostaciclina y óxido nítrico.
5. Expresión de difosfatasa de adenosina la cual hidroliza el difosfato de adenosina que es un agonista plaquetario.
6. En condiciones fisiológicas no expresa en su superficie moléculas de adhesión.
7. Regulación del tono arteriolar y del flujo de la microcirculación a través de la producción de óxido nítrico y prostaciclina.
La activación de las células endoteliales es fundamental en la
patogénesis de la sepsis ya que una vez son activadas por endotoxinas
y/o citocinas, amplifican la respuesta inflamatoria, el movimiento
célular (PMN), macrófagos, y la expresión de receptores de proteasa, los
cuales son activados por factor VIIIa, IXa y trombina. Una vez
activados inducen la síntesis en las células endoteliales de citocinas,
quimiocinas y moléculas de adhesión. (4)
 |
Asociado a este proceso, las células endoteliales
pierden trombomodulina y heparán sulfato, hay incremento en la síntesis
del FT, el cual impide la activación de la proteína C, el inhibidor del
FT y la ATIII, que asociado con la activación de la vía extrinseca por
la expresión del FT modifica el equilibrio procoagulante /anticoagulante
con franco predominio procoagulante. Esta respuesta fisiopatológica
modifica de manera significativa la microcirculación. Las células
endoteliales una vez activadas amplifican la respuesta inflamatoria, y
se inicia un círculo vicioso de inflamación, apoptosis, consumo de
proteína C, activación, disfunción y lesión endotelial, que evoluciona a
trombosis microvascular y disfunción órganica múltiple. (5)
Durante la activación de las células endoteliales se producen
micropartículas de fosfolípidos, que se caracterizan por ser pequeñas
estructuras vesiculares que transportan fosfatidilserina a la superficie
externa de la célula endotelial y que sirve de anclaje a los factores
de coagulación dependientes de vitamina K. Estas micropartículas
procoagulantes amplifican la vía extrínseca de la coagulación a través
del FT y favorecen la adhesión y agregación plaquetaria, además de dañar
irreversiblemente a la célula endotelial al ser mediadoras de
apoptosis. (6)
La Fibrinolísis se activa en fases iniciales del daño endotelial, lo
que incrementa los niveles del inhibidor del activador del plasminógeno,
lo que inhibe la fibrinólisis y trae como consecuencia que se acentué
más el desequilibrio procoagulante /anticoagulante.
Una vez que se inicia la infección / inflamación / coagulación /
disfunción endotelial, las células endoteliales, al estimularse expresan
en su superficie moléculas de adhesión, dentro de las que destacan:
P-selectina, E-selectina, molécula de adhesión intracelular y molécula
de adhesión vascular-1.
Los leucocitos interactuán con la célula endotelial y se inicia el
proceso de marginación, adhesión, rozamiento y transmigración que tiene
como finalidad la protección tisular, pero se torna nocivo dado que los
PMN activados liberan, enzimas proteolíticas y radicales libres de
oxígeno que amplifican el daño tisular y endotelial. (7)
Además de la activación y mal funcionamiento de las células
endoteliales durante la sepsis, el endotelio se lesiona, y se acentúa el
mal funcionamiento microvascular y el daño tisular. Los mecanismos
conocidos de daño endotelial están íntimamente relacionados al proceso
inflamatorio como sigue:
1. Los PMN activados se adhieren a las células endoteliales vía
moléculas de adhesión, producen lesión célular a través de radicales
libres y enzimas proteolíticas como la elastasa. Se ha demostrado en
modelos experimentales que el FNT alfa, potencia la acción tóxica de los
PMN. Este mecanismo de daño es importante en el paciente con sepsis
dado que productos de degranulación de neutrófilos como son la elastasa y
la lactoferrina incrementan sus niveles en presencia de FNT alfa y
están asociados a mal pronóstico.
2. Las citocinas principalmente FNT alfa y la IL-6 inducen apoptosis de las células endoteliales.
3. Linfocitos T citotóxicos y células naturales asesinas activadas por citocinas lesionan el endotelio vascular.
4. El mecanismo de isquemia-reperfusión a través de sus mediadores
como son: citocinas, complemento, neutrófilos y moléculas de adhesión,
disminuyen los niveles de ATP de las células endoteliales e inducen
apoptosis, además de amplificar la respuesta inflamatoria local.
5. La proteína C reactiva es un reactante de fase aguda estimulado
por IL-6 y usando como cofactor a la fosfolipasa A 2 que es una enzima
secretada por el endotelio dañado, activa el complemento. Los productos
del complemento activado a nivel endotelial amplifican la respuesta
inflamatoria y estimulan la síntesis del FT.
Existe una clara asociación entre coagulopatía y choque séptico, ya
que la respuesta inflamatoria que se presenta en sepsis, altera el
equilibrio procoagulante-anticoagulante y las propiedades
profibrinolíticas y anticoagulantes del endotelio vascular a
antifibrinolíticas y procoagulantes. (8)
La activación de la coagulación en sepsis grave es de etiología
multifactorial y la inducción de la expresión del FT a nivel endotelial
por la endotoxina es fundamental. Una vez expresado el FT se activa la
vía extrínseca de la coagulación que resulta en incremento en la
producción de trombina.
La trombina es una molécula de compleja actividad dado que además de su acción procoagulante tiene otras funciones como:
1. Inducción de la proliferación celular a través de la estimulación
de los siguientes mitógenos: Factor de crecimiento derivado de plaquetas
y el factor de crecimiento transformante Beta.
2. Amplifica la respuesta inflamatoria a través de mediar la expresión
de moléculas de adhesión y ser quimiotáctica directa de PMN, los cuales a
nivel tisular acentúan la lesión tisular por la liberación de enzimas
proteolíticas, fundamentalmente la elastasa, que tiene la capacidad de
inactivar al inhibidor de ATIII. (9)
La trombina se une a la trombomodulina, que es una de las proteínas
inhibidoras del estado procoagulable en la microcirculación. Esta
interacción bloquea la unión del fibrinógeno, plaquetas, y factor V a la
trombomodulina y a su vez el complejo trombina-trombomodulina activa a
la Proteína C. La proteína C activada (PCA) debe disociarse de su
receptor para interactuar con la proteína S y funcionar como
anticoagulante inactivando al factor Va. (10)
Existe una estrecha interrelación entre infección, lesión endotelial,
respuesta inflamatoria y coagulación. Citocinas inflamatorias como el
factor de necrosis tumoral y las interleucinas 1 y 6 son capaces de
activar la coagulación e inhibir la fibrinolísis. La trombina resultante
de la activación de la coagulación, además de su acción procoagulante
estimula la respuesta inflamatoria por múltiples vías. El resultado
final es daño endotelial generalizado, trombosis microvascular con
hipoxia e isquemia tisular y disfunción órganica múltiple. (11)
Referencia:
1. R. Phillip Dellinger. Inflammation and Coagulation Implications
for the Septic Patient. Clinical Infectious Diseases 2003;36: 1259-65
(15 May).
2. Bone RC. Grodzin CJ. Balk RA. Sepsis: a new hypothesis for pathogenesis of the diseases process. Chest 1997;112:235-43.
3. Gross PL. Aird WC. The Endothelium and thrombosis. Sem. Thromb Hemost 2000;26:463-478.
4. P. Marco I. Alberca Rev. Iberoamer Trom Hemostasia 2000;13(Sup. I):99-115.
5. Hack CE. Zeerleder S. The endothelium in sepsis: source of and a target for inflammation. Crit Care Medicine 2001;29:S21-7.
6. Aird WC. Vascular bed- specific hemostasis: role of endothelium in sepsis pathogenesis. Crit Care Medicine 2001;29:S28-34.
7. Tapper H. Herwald H. Modulation of hemostatic mechanism in bacterial infection diseases. Blood 2000;96:2329-37.
8. Dhainaut JF. Vallet B. Combined procoagulante and innate immune
responses to infection: toward more potent drugs in septic patients Crit
Care Medicine 2001;29:205-7.
9. Esmon CT. Does inflammation contribute to thrombotic events? Haemostasis 2000;30 (Suppl 2) :34-40.
10. Esmon CT. The endothelial cell Protein C receptor. Thromb Haemost 2000;83:639-43.
11. Vincent JL. Microvascular endothelial dysfunction: a renowed appreciation of sepsis pathophysiology. Crit Care 2001;5:S1-S5
Publicaciones relacionadas
- El ultrasonido musculoesquelético y la reumatología
- Evaluación pronóstica del uso de octreótide 2 vrs. 5 días en sangrado gastrointestinal superior de origen variceal
- Ley de Murphy para Médicos
- Acerca de nosotros y nuestros servicios
- Acromegalia
- Adyuvancia extendida en Cáncer de mama
- Aprendiendo, Jorge Luis Borges
- Areas nasales por tomografia axial computarizada:
- Ascitis: Mecanismos de producción. Diagnóstico y Tratamiento Dr. J. Alberto Marin
- Asociación CRECE
- Aspectos filosóficos e históricos en la práctica de la medicina interna Casi todos los médicos tienen sus enfermedades favoritas Dr. Hugo Raúl Castro Salguero. Médico Internista, Oncólogo. HGE – IGSS. Instituto Nacional de Cancerología INCAN. Grupo Médico Ángeles
- Calidad de vida en pacientes con hepatitis crónica por virus c vírgenes a tratamiento
- Canal: grupo medico angeles en youtube
- Características de los pacientes diagnosticados con enfermedad de Hansen en los últimos diez años en el INDERMA, Dr. Peter Greenberg Cordero, Director Médico del INDERMA; Dra. Suzzette de León, Jefa de Unidad de Hansen del INDERMA; Dra. Helga María Sarti, Residente III del INDERMA
- Carcinoma Basocelular Superficial: reporte de un caso
- Carcinoma de células de Merkel
- Caso Interesante
- Casos de Autodiscusión
- Casos de autodiscusión 2
- Cinco casos de enfermos con fiebre de origen desconocido
- Colonoscopia virtual 3d tan sensible como la colonoscopia de fibra
- Consejos útiles para dejar de fumar
- Cuerpo editorial
- Curiosidades de la Ciencia y de la Vida
- Curiosidades de la Ciencia y de la Vida 2